Bubrezi su vitalni organi koji čiste krv i održavaju ravnotežu tekućina u tijelu. Zovu ih i tihim organom, kod kojega smanjeni rad često ne daje specifične simptome dok bolest ne uznapreduje. Neke od bolesti teže je prepoznati, kako zbog nespecifičnih simptoma, tako i zbog nedostatka informacija. Do danas je poznato i više od stotinu različitih rijetkih bolesti koje zahvaćaju bubrege, a koje uključuju poremećaje razvoja, prijenosa tvari, metabolizma i upalne bolesti. Zbog različitih vidljivih osobina (fenotipske varijabilnosti), te malog broja poznatih bolesnika, definiranje bolesti, donošenje dijagnostičkih i terapijskih smjernica te pronalazak specifičnih lijekova za ove bolesnike često je dodatni izazov za liječnike.1
Dođe li do bilo kakvih oštećenja bubrega ili njihove funkcije, smanjuje se broj nefrona (osnovna jedinica građe bubrega koja filtrira krv). Kod smanjene količine nefrona, tijelo ne može pravilno održavati ravnotežu vode, tvari i elektrolita u organizmu, što može dovesti do niza zdravstvenih problema.
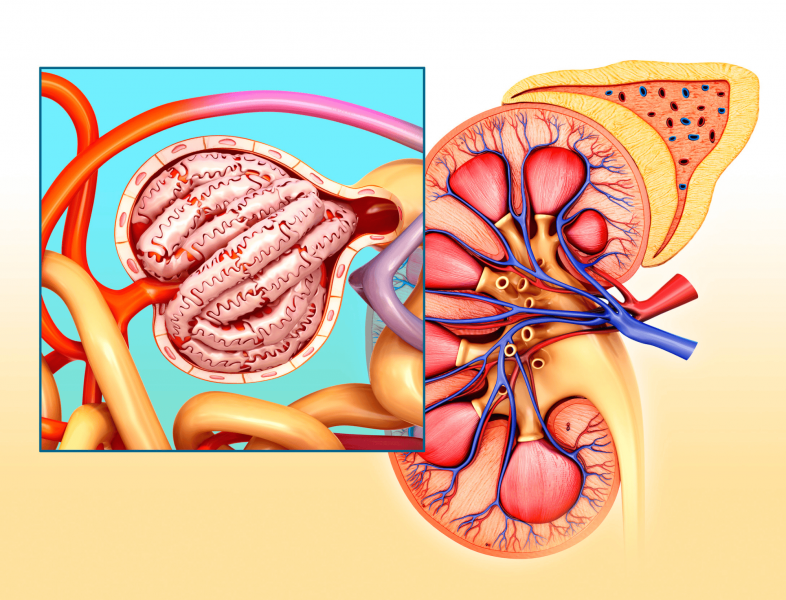
U nekim rijetkim bolestima bubrega, koje se nazivaju komplementom posredovane bolesti bubrega, oštećenje bubrega nastaje kada dio imunološkog sustava postane pretjerano aktivan.1-3
Što je komplementni sustav i zašto je važan za bubrege?
Komplementni sustav je dio urođene imunološke obrane i ima temeljnu ulogu u obrambenim mehanizmima protiv patogena kao što su virusi i bakterije. Sastoji se od proteina koji „surađuju“ kako bi uništili bakterije, viruse i druge štetne tvari. No, ako se ovaj sustav nekontrolirano aktivira, može uzrokovati oštećenja zdravih tkiva - osobito bubrega. U prvom redu su to atipični hemolitičko uremijski sindrom (aHUS) i C3 glomerulopatija (C3G), no sve je više znanstvenih dokaza koji podupiru teoriju aktivacije komplementa i u brojnim drugim bolestima bubrega kao što su različite vrste glomerulonefritisa, dijabetička nefropaija i lupus nefritis.2,3
Uloga sustava komplementa u raznim upalnim bolestima je višestruka; na primjer, aktivacija komplementa može uvelike pridonijeti upalom posredovanom oštećenju tkiva, dok nasljedni ili stečeni deficiti komplementa snažno pogoduju nastanku autoimunih bolesti. 5,7
Bubrezi su osjetljivi jer:
- sadrže gustu mrežu krvnih žila koje mogu izložiti tkiva djelovanju tih proteina
- nemaju dovoljno zaštitnih molekula koje bi ih obranile od vlastitog imunološkog napada
Iako rijetke, aHUS i C3 glomerulopatija mogu brzo napredovati do zatajenja bubrega. Rano prepoznavanje simptoma, posebice kod mlađih bolesnika s neobičnim nalazima u mokraći ili povišenim tlakom, može omogućiti bržu dijagnozu i bolju prognozu uz specijalizirano liječenje.2,5
Što je C3 glomerulopatija?
C3 glomerulopatija (C3G) je kompleksna, kronična, progresivna i ultra rijetka bolest bubrega, od koje obolijeva jedna do dvije osobe na milijun stanovnika.1,8 Češće se javlja u djece i adolescenata. Bolest je uzrokovana pretjeranom aktivnošću dijela komplementnog sustava - alternativnog puta komplementa, što dovodi do nakupljanja proteina C3 u bubrežnim filtarskim jedinicama - glomerulima.8
Postoje dva podtipa C3 glomerulopatija - dense deposit disease (DDD), kod kojih postoji gusto nakupljanje C3 proteina, te C3 glomerulonefritis (C3GN) kod kojega je nakupljanje C3 proteina blaže, ali svejedno dovodi do oštećenja bubrega. Oba podtipa uzrokovana su prekomjernom aktivacijom alternativnog puta komplementa, što dovodi do gubitka funkcije jednog od proteina koji reguliraju put komplementa (faktor H ili faktor I) ili stvaranja rezistencije na regulatorni faktor H.1,4,5
Zašto nastaje C3G?
Genske mutacije uzrok su bolesti u 10 do 20 posto slučajeva. Kod ostalih, za 50 do 80 posto najčešći uzrok C3 glomerulopacije jest nefritički faktor - autoprotutijelo koje stabilizira konvertazu, čime rezultira kroničnom aktivacijom komplementa i bubrežnim oštećenjem.1,4
Simptomi uključuju:
- krv u mokraći (hematurija),
- bjelančevine u mokraći (proteinurija),
- ponekad oticanje, povišeni tlak i smanjena bubrežna funkcija
Kako se dijagnosticira C3G?
Klinička slika uključuje proteinuriju i hematuriju uz očuvanu bubrežnu funkciju, nefrotski sindrom i rjeđe akutno bubrežno zatajivanje ili brzo progresivni glomerulonefritis.1-3 Svim bolesnicima sa proteinurijom i hematurijom potrebno je učiniti dijagnostičku obradu, koja uz laboratorijske testove uključuje i biopsiju bubrega. S obzirom na varijabilnu kliničku sliku, dijagnoza se postavlja isključivo na osnovu imunoflorescenije - mikroskopske metode pregleda tkiva.4
U svakog bolesnika koji se prezentira hematurijom i/ili proteinurijom, posebno uz znakove bubrežnog zatajivanja, hipertenzijom i niskim vrijednostima C3 možemo posumnjati na C3 glomerulopatiju. Kod mnogih oboljelih od C3 glomerulopatije s vremenom dolazi do zatajenja bubrega.4-6
Liječenje C3G
Danas imamo mogućnost ciljanog liječenja lijekovima koji djeluju na sam uzrok bolesti. Te bi terapijske opcije trebale usporiti napredovanje bolesti (smanjiti proteinuriju i stabilizirati ili poboljšati eGFR - procijenjenu brzinu filtracije bubrega), te spriječiti glomerularnu i tubulointersticijsku upalu, kao i bubrežnu fibrozu u bolesnika sa C3 glomerulopatijom.2,3 Takva terapija fokusirana je na blokiranje ranog dijela imunološkog sustava (komplementni sustav). Cilja specifičnu komponentu zvanu faktor B i time zaustavlja štetnu aktivaciju tog sustava.8 Na taj način sprječava daljnje oštećenje crvenih krvnih stanica i upalu koje su povezane s bolestima poput C3 glomerulopatije.5,7,8
Što je aHUS - atipični hemolitičko-uremijski sindrom
aHUS je rijetka, ozbiljna i progresivna bolest koja se javlja na otprilike 2-3 bolesnika na milijun stanovnika (2-3 : 1 000 000). Uzrokuje probleme u zgrušavanju krvi, razaranje crvenih krvnih stanica i oštećenje bubrega. Uglavnom se javlja u odraslih bolesnika, ali u 5-10 posto slučajeva i u pedijatrijskoj dobi.1,5
Zašto nastaje aHUS?
Najčešći uzrok je genetski poremećaj komplementnog sustava - mutacije različitih gena koji kodiraju komponente ili regulatorne proteine kaskade komplementa, odnosno smanjuju kontrolu nad aktivacijom komplementnog sustava.1,5 Ipak, iako osoba može naslijediti tu mutaciju, bolest se ne razvija kod svih nositelja, već su vjerojatno za oboljenje ključni i faktori iz okoliša, poput infekcija.5
Ponekad pak tijelo stvara protutijela koja napadaju vlastite zaštitne proteine (faktor H komplementa).
Simptomi uključuju:
- umor i blijedilo (zbog hemolitičke anemije),
- modrice i krvarenje (zbog trombocitopenije - manjka trombocita),
- povišeni krvni tlak,
- smanjena funkcija bubrega,
- u nekim slučajevima: neurološki simptomi, problemi sa srcem ili kožom
Kako se dijagnosticira aHUS?
Ne postoji test za aHUS, a dostupni biomarkeri ograničenog su značenja, tako da se bolest dijagnosticira metodom isključivanjem drugih uzroka. Klinička slika u pravilu uključuje tri karakteristična simptoma - hemolitičku anemiju, trombocitopeniju i razvoj zatajivanja bubrega.1,5 U 20 posto bolesnika razvijaju se i kardiovaskularni poremećaji (infarkt miokarda, zatajenje srca,..), neurološki (poremećaj svijesti, hemipareza – djelomična paraliza jedne strane tijela,..), plućni, gastrointestinalni ili dermatološki poremećaji.1,5,7 Sve to upućuje da je aHUS ponekad teško dijagnosticirati.
Kod sumnje na aHUS, važno je isključiti pristunost Shiga toksina (proizvode ga bakterije E. coli u zaraženoj hrani ili vodi), koji je najčešći uzrok aHUS-a i testirati vrijednost enzima ADAMTS 13 (koji je kod aHUS-a normalan, odnosno veći od 10 posto).1,5 Time razlučujemo aHUS od trombocitopenične purpure i infekcija.5
Preporučuje se učiniti testiranje koncentracije komplementa C3 i C4, te funkcije alternativnog puta CFH, CFI, protutijela na CFH i CFI te CH5 test aktivnosti komplementa, kako bi se utvrdilo je li riječ o atipičnom aHUS-u (uzrokovanom genetskim defektima ili autoantitijelima).5
Liječenje aHUS-a
Liječenje aHUS uključuje zamjenu bubrežne funkcije, plazmaferezu (pročišćavanje krvi), primjenu imunoglobulina i u novije vrijeme primjenu inhibitora komplementa, kao što su ekulizumab i ravulizumab.1,5
|
Kada potražiti liječničku pomoć?
|
Literatura:
- Thurman J.M. Complement in Kidney Disease: Core Curriculum 2015. Am J Kidney Dis.
2015;65(1):156-168. https://www.ajkd.org/article/S0272-6386(14)01189-5/fulltext - Morgan BP, Harris CL. Complement, a target for therapy in inflammatory and degenerative
diseases. Nat Rev Drug Discov. 2015 Dec;14(12):857-77. doi: 10.1038/nrd4657. Epub 2015
Oct 23. https://pubmed.ncbi.nlm.nih.gov/26493766/ - Luo W, Olaru F, Miner JH, Beck LH Jr, van der Vlag J, Thurman JM, Borza DB. Alternative
Pathway Is Essential for Glomerular Complement Activation and Proteinuria in a Mouse
Model of Membranous Nephropathy. Front Immunol. 2018 Jun 22; 9:1433.
doi:10.3389/fimmu.2018.01433. https://pubmed.ncbi.nlm.nih.gov/29988342/ - Vivarelli M, et al. The role of complement in kidney disease: conclusions from a Kidney
Disease: Improving Global Outcomes (KDIGO) Controversies ConferenceKidney
International (2024) 106, 369–391. https://pubmed.ncbi.nlm.nih.gov/38844295/ - Larosa F, et al. Complement System and the Kidney: Its Role in Renal Diseases, Kidney
Transplantation and Renal Cell Carcinoma. Int. J. Mol. Sci. 2023, 24, 16515.
https://doi.org/10.3390/ijms242216515 - West, E.E.; Kolev, M.; Kemper, C. Complement and the Regulation of T Cell Responses.
Annu. Rev. Immunol. 2018;3:309–338. https://pmc.ncbi.nlm.nih.gov/articles/PMC7478175/ - Leonardi, L, et al. Inherited Defects in the Complement System. Pediatr. Allergy Immunol.
2022, 33 (Suppl. 27), 73–76. https://pubmed.ncbi.nlm.nih.gov/35080299/ - Nester CM et al. Iptacopan reduces proteinuria and stabilizes kidney function in C3
glomerulopathy. KIReports 82025) 10. 432-446
https://www.sciencedirect.com/science/article/pii/S2468024924019892
Objavu članka sponzorirala je tvrtka Novartis Hrvatska d.o.o., broj odobrenja FA-11461793, 07/2025.